This resource has been developed to allow the comparison of CRISPRs between strains of a given species or between closely related species, and to classify the spacers. It is composed of two main applications.
When the genome sequence of several strains is available for a given species, and when each strain possesses several CRISPRs, it is important to be able to classify the different CRISPRs, particularly as their position on the genome might vary. A program has been created that automatically recovers from CRISPRdb all members of a genus containing a CRISPR and proposes to compare each of them. Additional strains can be added in the comparison. The comparison is based on the presence of identical DRs and similar flanking sequences (a certain mismatch threshold is accepted).
The result is shown in a table where CRISPRs are grouped. Information is given on their position and on the number of repeats. A link to the corresponding CRISPR in CRISPRdb can be activated.
For example in the genus Yersinia, and at the date of the writing of this tutorial, genome sequence data is available for six Y. pestis and two Y. pseudotuberculosis strains. Four different CRISPRs are found which can each be present in a subset of strains (in this particular case, one of the CRISPR loci is deleted in the leader region of the sequenced Y. pseudotuberculosis strains, and because of this feature is identified as a fourth locus).
When two or more alleles of a given CRISPR are found, the flanking sequences can be aligned and a link is provided to the second application "CRISPRtionary" to annotate and classify the spacers. This can be reached by activating the CompareSpacers button.
This is a very helpful tool to analyse sequences from multiple alleles derived from the same locus. Such data will be produced for instance when investigating the diversity (evolution) of CRISPRs within a species by sequencing the locus in different isolates. This tool can then be used to automatically number spacers, produce a "dictionary", and code the alleles using this dictionary.
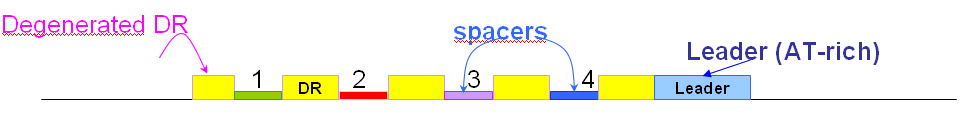
Several sequences can be submitted simultaneously in fasta format. CRISPRfinder identifies the consensus DR in each of them and extracts the spacer. In the next page a possibility is given to select the more appropriate DR. It is recommended to select the DR such that from left to right the degenerate DR is first and the leader is last. In this configuration the spacer 1 will be the first after the degenerated DR which corresponds to the oldest spacer inside the CRISPR.
Sample files are provided to illustrate how this works and what it does ( Y. pestis Dictionary and CRISPR_YP1_in Y. pestis).
The following steps need to be followed:
- Copy the sequences in the table or upload a file.
The sequences should be in fasta format, i.e for each sequence, the first line starts with the "greater than" symbol ">" and contains a unique identifier per sequence. This is the sequence header and must be in a single line. It is possible to put additional fields in the header separated by a pipeline "|", these fields will be especially useful in the final output files.
If a catalogue of annotated spacers is already available for the analysed species, it is recommended to use this catalogue as a spacers dictionary. The proper catalogue format is an excel file with the following properties :
- the first row should contain columns labels
- the first column should contain the spacer labels
- the second column may contain alternative labels or information about the spacers
- the third column should contain the spacers sequences
- the first three columns should not be empty and should not contain skipped rows.
In a single excel file, information on different CRISPRs can be stored in different sheets.
(you can easily get a proper sample catalogue file by running an analysis without loading a catalogue).
- Activate the Find Crisprs button
If a dictionary was uploaded, select the sheet in which the CRISPR information is present.
If no dictionary was uploaded, just go through the next step and a dictionary will be created.
At this step several DRs might be proposed and you have to select one that will be used to identify the spacers. It should be such that the orientation of the CRISPR is as explained above i.e. the leader on the right and the diverged DR on the left.
It is possible to allow the existence of mismatches between spacers.
- Activate the Find Spacers button
Here the DR and spacers are separated and each spacer is given a number. If a dictionary was uploaded only newly identified spacers are given a new number (in sequential order).
As the spacer numbering will only depend on the order in which the alleles are analysed it may be useful to re-annotate the spacers to produce a more logical numbering. This can be done by using the re-annotate Spacers function either on the initial excel file or after new alleles are analysed.
When re annotating, only spacers marked with a number will be considered. The presence of a letter in the spacer Id prevents re-annotation.
As an output of the spacers extraction and numbering, different files can be recovered:
- AnnotFasta:
a text file representing the corresponding CRISPRs. Each motif (DR+ spacer) is written on a separate line, the DR and spacer are separated by a tabulation and followed by the spacer label.
- AnnotFasta_CodedAlleles:
the same file as the previous one in addition to the spacers codes in the header separated by dots.
- Fasta_CodedAlleles:
the previous file represented in fasta format.
- Table_Coded_Alleles:
excel file representing one allele per row. The header informations (separated by a pipeline in the submitted sequences) are presented in separate columns. The last column provides the spacers labels separated by dots.
- Initial dictionary:
the initial uploaded dictionary
- New dictionary:
the updated dictionary
- binary file:
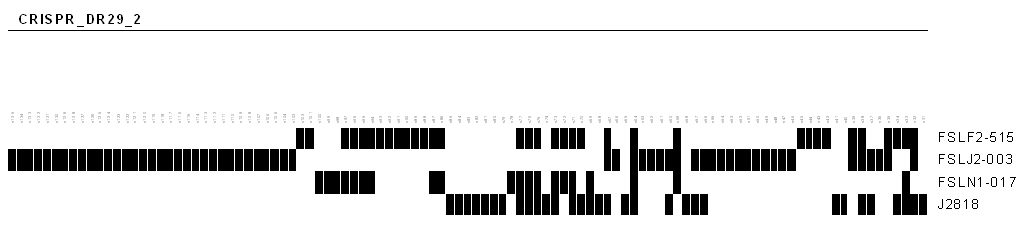
excel file where columns represent the spacer labels and rows represent the queried alleles. For each CRISPR allele, spacer will be given the \931\94 value when it exists and 0 when it is absent. The binary file is especially interesting for providing a spoligotyping profile of the CRISPR and to visually illustrate the spacers composition in the strains as illustrated for instance in the following figure.
 |