CRISPRs Web service User's manual
Summary
A number of tools are available here:
- The CRISPR database itself, in which microbial genomes have been pre-processed in search for CRISPRs structures. This database can be browsed in an intuitive way.
-
The second tool is the crispr finder tool. You can use this page to analyse your own data.
-
The third utility provides a global overview of CRISPRs present in the database. Lists of known DRs and spacers, and of CRISPRs ordered according to the number of spacers, are provided, with links to detailed information for the corresponding CRISPRs. Sequence alignments of selected DR regions can be produced using CLUSTAL W, together with dendrograms.
-
The BLAST CRISPR page will be of use to try to validate a questionable CRISPR. From this page, a candidate DR region (or spacer) can be compared to all DRs (or spacers) characterised so far from clear-cut CRISPR structures present in the database.
-
The FlankAlign link is useful to compare CRISPR flanking sequences, for
instance when looking for the homolog of a CRISPR locus in other strains,
or when trying to validate a questionable CRISPR, by searching for a
leader sequence.
-
MyCRISPRdb allows you to store your own data. Your data will be available together with that of the main database for comparison with the CRISPRcompar tool.
-
The last resource is the "spacers dictionary". This is a very helpfull tool to analyse sequence from multiple alleles derived from the same locus. Such data would be produced for instance when investigating the diversity of CRISPR within a species by sequencing the locus in different isolates. This tool can then be used to automatically number spacers, produce a "dictionary", and code the alleles using this dictionary. Sample files are provided to illustrate how this works and what it does ( Pestis Dictionary and CRISPR_YP1_Pestis).
The first database page displays the list of prokaryotic public genomes (stored in the database and updated frequently). The species may be displayed in two ways :
- a) View the strains alphabetical browser
this link shows the list of strains in the alphabetical order. The bacterial and the archaeal lists are separated.
- b) View the strains taxonomy browser
this link shows the strains list according to taxonomic rank. Five taxonomic ranks are allowed.
The colour code indicates whether a CRISPR has been detected or not: strains without a CRISPR are coloured in yellow, strains having at least one CRISPR are coloured in pink and strains having only questionable CRISPRs are in orange.
Upon selecting a strain name, a page displays the strain properties, the available genomes (chromosome and plasmids) and indicates how many CRISPRs have been found. The colour code is the same as above.
The button  leads to the CRISPRs properties page giving more details on the found CRISPRs. The button Find cas genes leads to a table showing the list of CRISPR-associated genes annotated in all the genomes (plasmids or chromosomes) in the corresponding taxon and their position on the genome.
leads to the CRISPRs properties page giving more details on the found CRISPRs. The button Find cas genes leads to a table showing the list of CRISPR-associated genes annotated in all the genomes (plasmids or chromosomes) in the corresponding taxon and their position on the genome.
Example
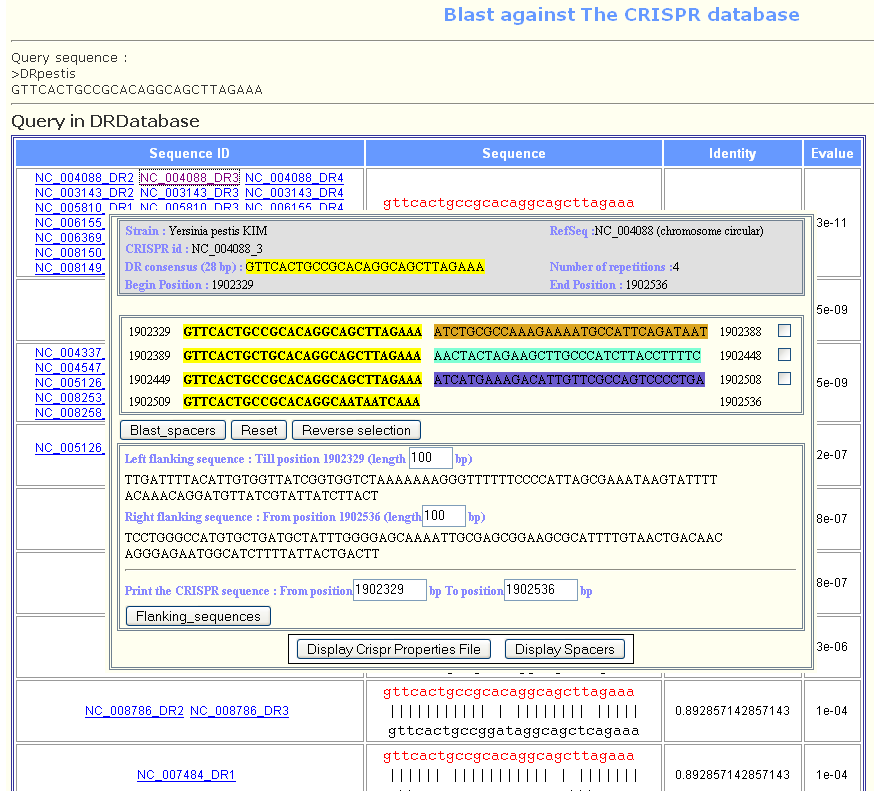
The CRISPRs properties page, indicates the CRISPR's id together with its position on the genome, the number of spacers and the consensus DR sequence.
Querying a CRISPR locus leads to a page containing all its properties : the DR consensus shown in yellow, the spacers shown in different colours, their positions in the genome.
- The left flanking sequence is given by default. It is the sequence that ends at the begin position of the CRISPR and has a specified length (100 bp by default).
The user may change this value to get longer or shorter sequences by modifying the length value
and then clicking on flanking_sequences button.
- The right flanking sequence is given by default. It is the sequence that begins from the end position of the CRISPR and has a specified length (100 bp by default). The user may change this value to get longer or shorter sequences by modifying the length value and clicking on flanking_sequences button.
- CRISPR sequence is the sequence from the first nucleotide in the first DR to the last nucleotide of the last DR. To get this sequence or to modify its start or end positions, the user should click on the flanking sequence button.

This page provides a global overview of CRISPRs present in the database and offers the possibility of downloading some overview files.
- Exhaustive DR List and DRs alignement
DRs belonging to confirmed CRISPRs are listed. The corresponding ids of each DR are also provided for further exploration of the related CRISPRs by clicking on the id.
Multiple sequence alignment of selected DR regions is obtained by clicking on the button Align DR sequences. Alignment is produced using default parameters of ClustalW program and the dendrogram is drawn using the ".dnd" file generated by this program.
- View and blast repeated Spacers list
The list of spacers reports the spacers which are found more than once in the database (exactly the same). By checking the blast box, user can blast the selected spacer againts All GenBank+EMBL+DDBJ+PDB sequences (but no EST, STS). The displayed results meet the following criteria :
- E-value <= 0.1
- the hit sequence size >= 70% of the queried sequence size
- Number of spacers in each CRISPR
This table provides the list of CRISPRs in bacteria and archeae sorted by number of spacers. So, it gives an idea about the number of spacers in the CRISPRs of the database and indicates the longest and the shortest ones.
- Download files
It is possible to download text files with the following information :
- Exhaustive DR list. Only DRs of confirmed CRISPRs are reported,
- Exhaustive Spacers list. Only spacers of confirmed CRISPRs are reported,
- List of genomes having confirmed CRISPRs,
- List of genomes without CRISPRs.
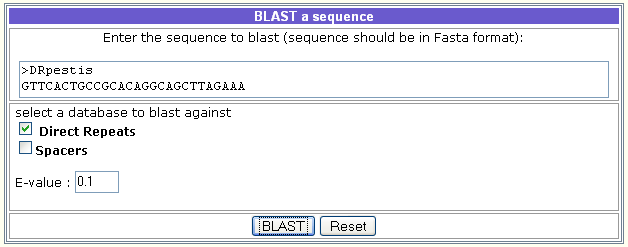
The BLAST against the CRISPRdb database finds regions of local similarity between the introduced nucleotide sequence(s) and the catalogue of DRs or (and) spacers of confirmed CRISPRs.
It is used as follows:
- Introduce the query sequence and select Direct Repeats or (and) Spacers.
- The result is displayed in the next page and for more details, user may click on the sequence id to display more information about the related CRISPR.
- Purpose of the page
This page is conceived for aligning CRISPRs flanking sequences. It is especially useful for identifying and comparing the leader sequences. The comparison may be done for CRISPRs of the same genomic sequence or for different genomic sequences, for example, comparing leaders of CRISPRs having the same DR or comparing leaders of a circular chromosome and a leader in a plasmid, etc.
- Example of use
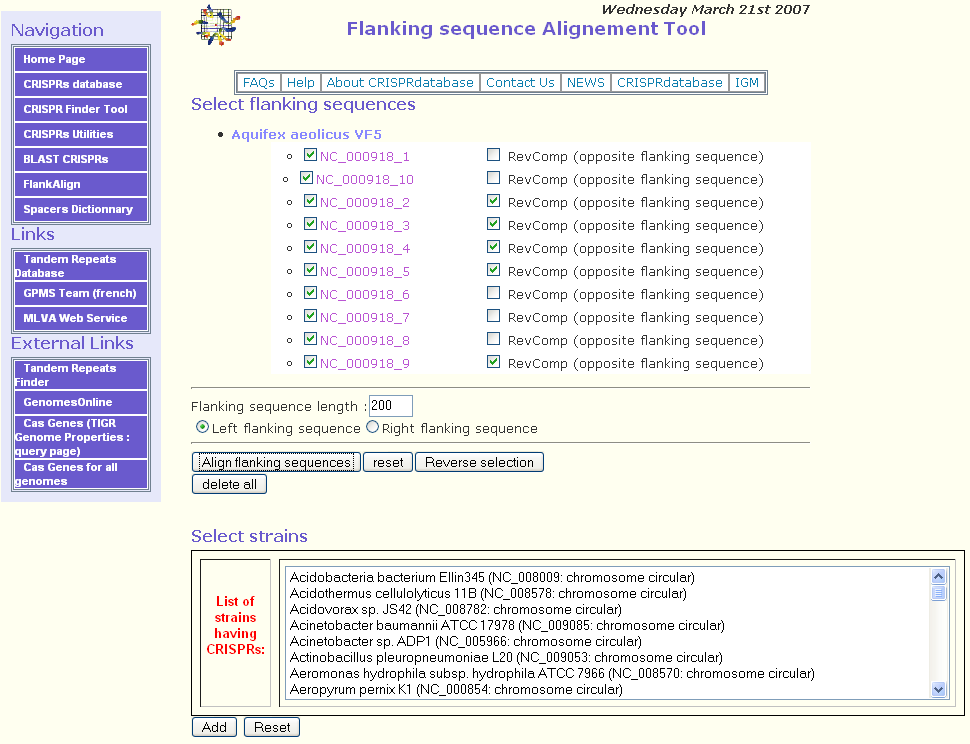
As an example, we have chosen to align the leader sequences of the CRISPRs of the circular chromosome of Aquifex aeolicus VF5 (RefSeq = NC_000918). In this genomic sequence, there are ten CRISPRs having a similar DR as shown in CRISPRdb database.
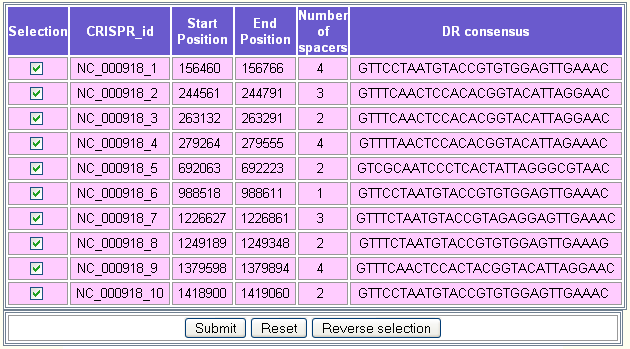
Five of them (NC_000918_1, NC_000918_6, NC_000918_7, NC_000918_8, NC_000918_10) are on the direct strand and five (NC_000918_2, NC_000918_3, NC_000918_4, NC_000918_5, NC_000918_9) are on the reverse complement strand.
- i)
The first step consists in selecting one or several genomic sequences to analyze. Only the genomic sequences having at least one CRISPR are displayed.
In this example, we selected the circular chromosome of Aquifex aeolicus VF5 (RefSeq = NC_000918). Ten putative CRISPRs will be displayed as shown in the following figure.
- ii)
The second step consists in selecting the CRISPRs for which the user would like to align the flanking sequences. By default, all the CRISPRs are selected. The button Reverse selection may be used to deselect them.
Once the CRISPRs are selected, the user should specify the flanking sequence length for the alignment. The default parameter is 200 bp. After that, the right or the left flanking sequence should be selected. By default, the left flanking sequence is checked. In fact, when the CRISPR is on the direct strand, the leader sequence is on the left.

If some CRISPRs are on the reverse complement strand, checking the checkbox RevComp (opposite flanking sequence) will align the opposite flanking sequence of the related CRISPR. As you can see in the figure above, for CRISPRs NC_000918_2, NC_000918_3, NC_000918_4, NC_000918_5, NC_000918_9 this option was selected because these CRISPRs are on the reverse complement strand.
- iii)
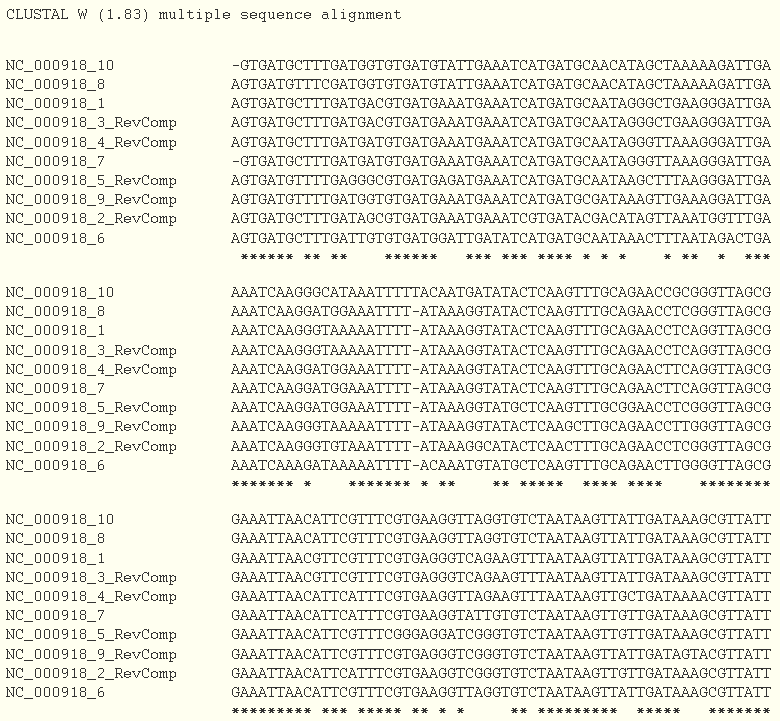
The last step consists in checking the Align flanking sequences button. A multiple alignment will be done using the ClustalW program.
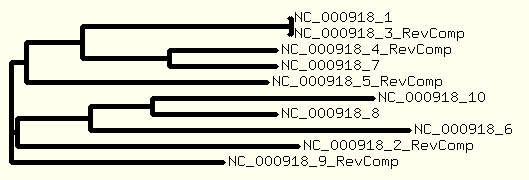
And a tree is drawn based on the ".dnd" file generated by ClustalW program.
- Create your login
First register by entering your email address and password, then activate the register button. Login to access the Private connection.
- Find and store your CRISPRs
Submit your own data directly in the box or upload a file. The FindCRISPR button will activate CRISPR search using default parameters. The advanced version allows you to modify the parameters for Maximal repeats and CRISPR properties.
The result of the query is displayed in a table. The number of confirmed and questionable CRISPRs is shown.
At the bottom of the page, enter the name of your sequence and save in the private database.
- Your private space
Two actions are offered within your Private space:
- Consult your private database to access the data stored in it. The CRISPRs are displayed as described in the main database section .
- Make a CRISPR comparison to compare CRISPRs which are present in the main database and in your private database (see the CRISPRcompar section).
The Spacer Dictionnary Creator use is illustrated by an example based on Yersinia pestis sample data (see related paper ).
The user should introduce:
- a dictionnary file which is an excel file containing a sheet with three columns in the currently used format (will be reduced to two in the future): a column for the spacer id, a column for alias names (may be empty) and a column for the spacer sequence (as an example see the file).
The tool can be run without loading any dictionnary file.
- a fasta file with multiple alleles sequences (see the example).
Next, the user has to select the sheet (of the excel dicitonnary to be used in the analysis.
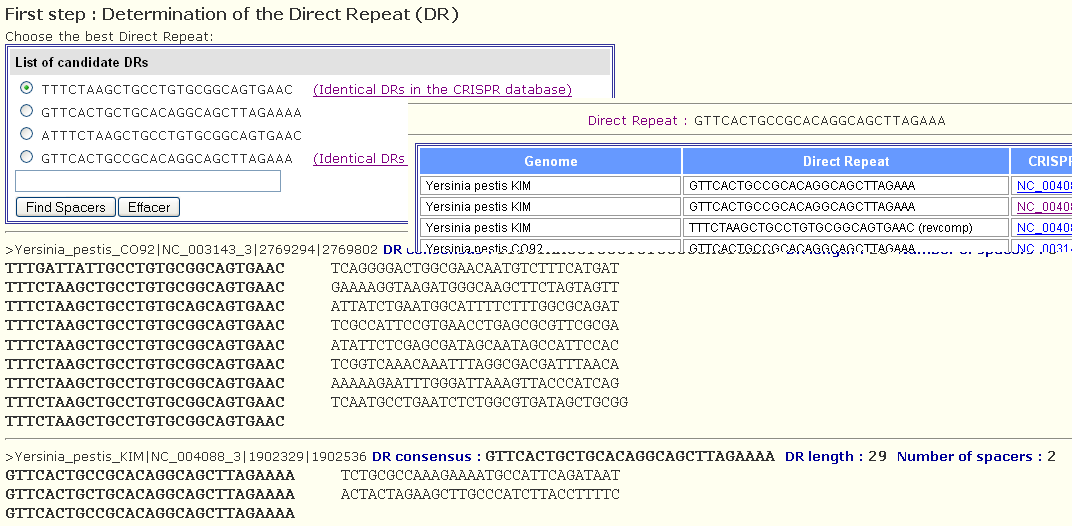
Then, each sequence of the fasta file will be analyzed by CRISPRFinder and the related CRISPR will be detected. In some cases, no CRISPR is detected because the Direct Repeat sequences are too diverged but this problem will be treated in the next step.
As the sequences are generally short or may contain some sequencing errors, the DR of each cluster may be not defined accurately in this step, so the user should select manually one DR among the obtained DRs listed as shown by the following figure. When identical DRs are detected in the database, a link for more details on it appears next to the DR sequence. This may help the user in selecting the appropriate DR sequence.
Finally, the selected DR will be blasted against all the introduced sequences and all the CRISPRs will be shown (even degenerated ones that did not appear in the previous page). An id will be assigned to each spacer. The id is either selected from the dictionnary (when the spacer is already in the dictionnary) or assigned a number and added to the dictionnary (if it is not listed in the dictionnary).

The colour code indicates whether a CRISPR has been detected or not:
- a)Yellow
No CRISPRs found,
- b)Pink
Confirmed CRISPR,
- c)Orange
Questionable CRISPR.
Species without a CRISPR are coloured in yellow, species having at least one CRISPR are coloured in pink and species having only questionable CRISPRs are in orange.
There are two kinds of "questionable" CRISPRs:
- Small CRISPRs, i.e structures having only two or three DRs
- Similar structures like particlar kinds of tandem repeats (not eliminated) or structure where the repeated motifs (DR in CRISPR) are not 100% identical.
They stop being questionable if the DR consensus is found elsewhere in the database in a convincing CRISPR.
Many of these structures are not true CRISPRs, and they need to be critically investigated. One way to "critically investigate" is to see if
the questionable CRISPR seems to be within a coding sequence. CRISPR are
usually non-coding, and do not belong to genes. An other way is to check
the internal conservation of the candidate DRs, and the divergence of the
candidate spacers. More definitive evidence might be provided by the
typing of a collection of strains from this species. Some bench work is
needed there.